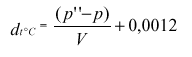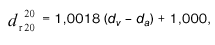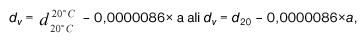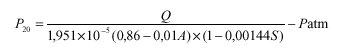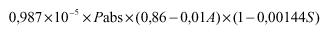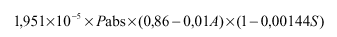Na podlagi 10. točke 42. člena zakona o vinu in drugih proizvodov iz grozdja in vina (Uradni list RS, št. 70/97 in 16/01) izdaja minister za kmetijstvo, gozdarstvo in prehrano
NAVODILO
o fizikalno kemijskih analizah grozdnega mošta in vina
1. člen
(fizikalno kemijske analize)
Ta navodilo določa postopke in aparature za naslednje fizikalno kemijske analize:
– določanje gostote in relativne gostote;
– določanje alkohola;
– določanje skupnega suhega ekstrakta;
– določanje skupnih kislin;
– določanje hlapnih kislin;
– določanje nehlapnih kislin;
– določanje pepela;
– določanje pH vrednosti;
– določanje reducirajočih sladkorjev;
– določanje prostega in skupnega žveplovega dioksida;
– določanje sladkorja z refraktometrijo;
– določanje saharoze;
– določanje citronske kisline;
– določanje ogljikovega dioksida.
Za analizo vina in mošta se lahko uporabljajo tudi druge metode, katerih rezultat mora biti enak rezultatom metod, ki jih določa to navodilo.
2. člen
(odločba o ocenitvi)
Analize vina in mošta izvajajo pooblaščene organizacije, ki izdajo odločbo o ocenitvi v upravnem postopku. V primeru nestrinjanja naročnika ocenitve z odločbo morajo pooblaščene organizacije pri ponovni ocenitvi uporabiti le tiste metode, ki jih določa to navodilo.
I. Določanje gostote in relativne gostote pri 20 °C
3. člen
(splošno)
Gostota je masa vina ali mošta na enoto volumna pri 20 °C. Izražena je v gramih na mililiter in označena s simbolom d(20).
Relativna gostota pri 20 °C je razmerje med gostoto določenega volumna vina ali mošta pri 20 °C in enakega volumna vode pri enaki temperaturi. Označena je s simbolom d(20)(na 20).
Gostoto in relativno gostoto pri 20 °C izmerimo na testnem vzorcu z uporabo referenčne metode (piknometrija) ali z uporabo dovoljene metode (hidrometrija ali denzimetrija z uporabo hidrostatske tehtnice).
Za zelo natančno merjenje je potrebna korekcija gostote zaradi vpliva žveplovega dioksida po formuli:
d(20)= d'(20) - 0,0006 x S
kjer pomeni:
d(20) korigirana gostota;
d'(20) navidezna gostota;
S koncentracija skupnega žveplovega dioksida v gramih na liter.
Če vino ali mošt vsebuje znatne količine ogljikovega dioksida, predpripravo vzorca izvedemo tako, da večino ogljikovega dioksida odstranimo z mešanjem 250 ml vina v litrski bučki ali s filtracijo pod zmanjšanim pritiskom (vakuumsko filtracijo) skozi 2 g bombažne vate, ki jo damo v razširjeno epruveto.
4. člen
(referenčna metoda)
a) Aparature: normalna laboratorijska oprema, posebno pa:
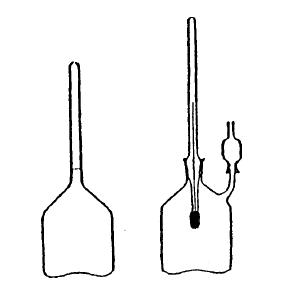
– Piknometer: iz Pyrex stekla, volumna približno 100 ml in s snemljivim termometrom, ki je s piknometrom povezan z obrusom in je kalibriran v desetinkah stopinje od 10 do 30 °C (slika 1). Termometer mora biti standardiziran. Piknometer ima 25 mm dolgo stransko cevko z maksimalnim notranjim premerom 1 mm, ki se končuje s koničnim obrusom. Ta stranska cevka je lahko pokrita z zamaškom, ki ima prav tako koničen obrus in se končuje z vodoravnim prerezom. Zamašek služi kot raztezna celica. Oba obrusa aparature morata biti narejena zelo pazljivo.
– Posoda tara: sestavljena iz piknometra (2), ki naj ima enak zunanji volumen kot piknometer (1) (razlika volumna sme biti največ 1 ml) in z maso enako masi piknometra (1), napolnjenega s tekočino s specifična maso 1,01 (2,0% raztopina natrijevega klorida).
– Toplotno izolirana posoda, ki se točno prilega piknometru.
– Dvoramenska (twin-pan) tehtnica z merilnim območjem najmanj 300 g in možnostjo odčitavanja na 0,1 mg natančno ali enoramenska (single pan) tehtnica z merilnim območjem najmanj 200 g in možnostjo odčitavanja na 0,1 mg natančno.
Slika 1: Piknometer in posoda tara
b) Umerjanje piknometra
Umerjanje piknometra vključuje določevanje naslednjih vrednosti:
– maso praznega piknometra,
– volumen piknometra pri 20 °C,
– maso piknometra napolnjenega z vodo pri 20 °C.
1. Metoda z uporabo dvoramenske (twin-pan) tehtnice:
Tara posodo postavimo na levo stran tehtnice in čist ter suh piknometer, opremljen z zamaškom, na desno stran tehtnice. Posodi, ki drži piknometer, dodamo uteži in zapišemo maso, ki je potrebna za ravnovesje; to maso označimo s p gramov.
Piknometer pazljivo napolnimo z destilirano vodo pri sobni temperaturi in pritrdimo termometer. Piknometer pazljivo obrišemo, ga osušimo in postavimo v toplotno izolirano posodo. Mešamo z obračanjem posode, dokler temperatura, ki jo odčitamo na termometru, nima konstantne vrednosti. Na vrhu stranske cevke točno uravnamo nivo vode, nato jo osušimo (obrišemo) in zapremo z zamaškom. Pazljivo odčitamo temperaturo t (v °C) in pri tem upoštevamo korekcijo netočnosti temperaturne skale. Stehtamo z vodo napolnjen piknometer “proti” tara posodi in zapišemo maso p' v gramih, ki je potrebna za vzpostavitev ravnotežja.
Izračun:
– Tariranje praznega piknometra:
masa praznega piknometra = p + m,
kjer je m masa zraka v piknometru: m = 0,0012 (p – p')
– Volumen pri 20 °C:
V (20 °C)= (p + m – p') × F(t), kjer je:
F(t)= faktor (tabela I, ki je v prilogi 1 objavljena skupaj s tem navodilom, kot njegov sestavni del) za temperaturo t °C,
V (20 °C) mora biti podan na + / - 0,001 ml natančno.
– Masa vode pri 20 °C:
M (20 °C)= 0,998203 V (20 °C), kjer je:
0,998203 gostota vode pri 20 °C.
2. Metoda z uporabo enoramenske (single-pan) tehtnice:
Določimo:
– maso čistega in suhega piknometra; to označimo s P,
– maso piknometra, napolnjenega z vodo pri t °C, kot je opisano v b) 1. točki tega člena; to označimo s P(1),
– maso tare; to označimo s T(0).
Izračun:
– Tariranje praznega piknometra:
masa praznega piknometra = P – m,
kjer je m masa zraka v piknometru: m= 0,0012 (P(1) – P).
– Volumen pri 20 °C:
V (20 °C)=(P(1) – (P – m)) × F(t), kjer je:
F(t)= faktor (iz tabele I v prilogi 1) za temperaturo t °C;
Volumen pri 20 °C mora biti podan na + / - 0,001 ml natančno.
– Masa vode pri 20 °C:
M (20 °C)= 0,998203 V (20 °C), kjer je:
0,998203 gostota vode pri 20 °C.
c) Metode merjenja:
1. Metoda z uporabo dvoramenske (twin-pan) tehtnice
Piknometer napolnimo s pripravljenim vzorcem po navodilih iz b) 1. točke tega člena.
p''= masa (v gramih), potrebna za vzpostavitev ravnotežja pri t °C,
masa tekočine v piknometru= p + m – p''.
Navidezna gostota pri t °C:
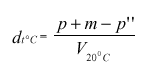
Za izračun gostote pri 20 °C uporabimo ustrezno korekcijsko tabelo, glede na vrsto merjenega vzorca: za suho vino uporabimo tabelo II, ki je v prilogi 1, za naravni ali koncentrirani mošt uporabimo tabelo III, ki je v prilogi 1, za sladko vino uporabimo tabelo IV, ki je v prilogi 1.
Relativno gostoto vina ali mošta izračunamo:
kjer je d(20) gostota vina pri 20 °C (korigirano na prosti in skupni žveplov dioksid) v gramih na liter in 0,998203 gostota vode pri 20 °C v gramih na liter.
2. Metoda z uporabo enoramenske (single-pan) tehtnice
Stehtamo tara posodo; to maso označimo s T;
izračunamo: DeltaT= T(1) – T(0);
masa praznega piknometra v času merjenja = P – m +DeltaT;
stehtamo piknometer, napolnjen s pripravljenim vzorcem, kot je opisano v b) 1. točki tega člena; njegovo maso pri t °C označimo s P(2).
Masa tekočine v piknometru pri t °C = P(2) – (P – m + DeltaT);
navidezna gostota pri t °C:
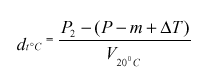
Izračunamo gostoto tekočine pri 20 °C (suho vino, naravni ali koncentrirani mošt, sladko vino), kot je opisano v c) 1. točki tega člena.
Relativno gostoto vzorca izračunamo tako, da delimo njegovo gostoto pri 20 °C z 0,998203.
3. Ponovljivost merjenja gostote (r):
– za suha in polsuha vina: r = 0,00010,
– za sladka vina: r = 0,00018.
4. Obnovljivost merjenja gostote (R):
– za suha in polsuha vina: R = 0,00037,
– za sladka vina: R = 0,00045
5. člen
(običajne metode)
1. HIDROMETRIJA
a) Aparature:
– Hidrometer:
Hidrometri morajo po dimenzijah in razdelitvi skale ustrezati ISO standardom.
Imeti morajo cilindrično bučko in cevko s krožnim prerezom minimalnega premera 3 mm. Za suha vina morajo biti umerjeni od 0,983 do 1,003 z razdelki 0,0010 in 0,0002. Vsak razdelek pri 0,0010 mora biti ločen od naslednjega vsaj za 5 mm. Za meritve specifična mase dealkoholiziranih vin, sladkih vin in moštov uporabljamo set petih hidrometrov, umerjenih od 1,000 do 1,030, od 1,030 do 1,060, od 1,060 do 1,090, od 1,090 do 1,120 in od 1,120 do 1,150. Ti hidrometri naj bodo umerjeni v enotah gostote pri temperaturi 20 °C, z razdelki vsaj na vsakih 0,0010 in 0,0005, vsak razdelek pri 0,0010 naj bo vsaj 3 mm ločen od naslednjega.
Hidrometri morajo biti umerjeni tako, da odčitavamo “na vrhu meniska”. Oznaka o umeritvi gostote ali relativne gostote in o odčitavanju “na vrhu meniska” mora biti na umeritveni skali ali na kosu papirja, zaprtem v bučki. Hidrometre umerja s strani države pooblaščena institucija.
– Umerjen termometer, ki ima skalo z minimalnimi razdelki 0,5 °C.
– Merilni valj, z notranjim premerom 36 mm in višino 320 mm, postavljen vertikalno s pomočjo izravnalnih vijakov.
b) Postopek
– Metoda merjenja:
250 ml testnega vzorca nalijemo v merilni valj (tretja alinea 1.a) točke tega člena) in vanj vstavimo termometer in hidrometer. Temperaturo odčitamo po enominutnem mešanju vzorca, s katerim izenačimo temperaturo. Termometer umaknemo in odčitamo navidezno gostoto pri t °C s skale hidrometra po preteku naslednje minute.
– Navidezno gostoto pri t °C korigiramo na 20 °C z uporabo pripadajočih tabel: za suha vina (tabela V, ki je v prilogi 1), za mošte (tabela VI, ki je v prilogi 1) in za sladka vina (tabela VII, ki je v prilogi 1).
– Relativno gostoto vina ali mošta izračunamo tako, da delimo njegovo gostoto pri 20 °C z 0,998203.
2. DENZIMETRIJA Z UPORABO HIDROSTATSKE TEHTNICE
a) Aparature:
– Hidrostatska tehtnica z maksimalno kapaciteto vsaj 100 g in možnostjo odčitavanja na 0,1 mg natančno.
– Dva identična plovca iz Pyrex stekla z volumnom najmanj 20 ml. Pritrdimo jih pod obe posodi tehtnice tako, da jih obesimo s pomočjo žice z maksimalnim premerom 0,1 mm.
Plovec, obešen pod desno posodo tehtnice, moramo vstaviti v merilni valj z oznako, ki označuje nivo. Merilni valj mora imeti najmanj 6 mm večji notranji premer, kot je premer plovca. Plovec mora biti popolnoma potopljen v merilnem valju pod oznako, gladina merjene tekočine je lahko prekinjena le z žico, na katero je obešen plovec. Temperatura merjene tekočine v merilnem valju je izmerjena s termometrom, ki je umerjen na 0,2 °C natančno.
– Uporabimo lahko tudi enoramensko (single-pan) tehtnico.
b) Postopek:
– Standardizacija hidrostatske tehtnice:
Z obema plovcema v zraku vzpostavimo ravnotežje z dodajanjem uteži v desno posodo; maso dodanih uteži označimo s p.
Merilni valj napolnimo z vodo do oznake, ga pretresemo in odčitamo temperaturo t °C po dveh do treh minutah.
Ponovno vzpostavimo ravnotežje s pomočjo uteži, ki jih dodamo v desno posodo; maso dodanih uteži označimo s p'.
Volumen plovca pri 20 °C je podan z enačbo:
V (20°C) = (p' – p) (F + 0,0012);
Faktor F za temperaturo t °C je podan v tabeli I v prilogi 1, p in V(20 °C) pa sta karakteristična za plovec.
– Metoda merjenja:
Desni plovec potopimo v merilni valj z vinom (ali moštom) do oznake. Odčitamo temperaturo t °C vina (ali mošta) in maso uteži, ki jo označimo s p''. Potrebujemo jo, da vzpostavimo ravnotežje.
Navidezna gostota d(t°C) je podana s formulo:
Če je plovec iz Pyrex stekla, navidezno gostoto korigiramo na 20 °C z uporabo tabel II, III ali IV, ki so v prilogi 1.
II. Določanje alkohola
6. člen
(splošno)
Volumenski delež alkohola pomeni število litrov etanola, ki ga vsebuje 100 litrov vina; oba volumna sta merjena pri 20 °C. Enota so %.
Upoštevamo, da so homologi etanola, skupaj z etanolom in homologi etanola v etilnih estrih, všteti v volumenskem deležu alkohola, ker so prisotni v destilatu.
Principi metod:
1. Metoda za pridobivanje destilata: destilacija vina, ki je naalkaljeno s suspenzijo kalcijevega hidroksida; merimo volumski delež alkohola v destilatu.
2. Referenčna metoda: merjenje gostote destilata s piknometrom.
3. Dovoljene metode:
– Merjenje volumskega deleža alkohola v destilatu s hidrometrom,
– Merjenje volumskega deleža alkohola v destilatu z denzimetrijo ob uporabi hidrostatske tehtnice,
– Merjenje volumskega deleža alkohola v destilatu z refraktometrijo.
Upoštevamo, da se za preračunavanje volumskega deleža alkohola iz gostote destilata uporabljajo tabele I, II, III, ki so v prilogi 2 objavljene skupaj s tem navodilom, kot njegov sestavni del.
Tabela I, ki je v prilogi 2, prikazuje splošno formulo, ki povezuje volumski delež alkohola z gostoto mešanic alkohol-voda kot funkcijo temperature.
7. člen
(metoda za pridobivanje destilata)
a) Aparature:
– Destilator, ki je sestavljen iz okrogle bučke, volumna 1 liter z obrusom; približno 20 cm visoke rektifikacijske kolone ali katerekoli aparature, ki prepreči brizganje; vira toplote (piroliza ekstrakta mora biti preprečena s primernimi ukrepi) in hladilnika s cevjo, ki destilat vodi na dno umerjene bučke, ki vsebuje nekaj mililitrov vode.
– Parni destilator, ki je sestavljen iz razvijalca pare, cevi za paro, rektifikacijske kolone in hladilnika.
Uporaben je katerikoli tip destilatorja ali parnega destilatorja, ki ustreza sledečemu testu:
Destiliramo zmes vode in etanola z volumenskim deležem 10%, petkrat zapovrstjo. Destilat mora vsebovati najmanj 9,9% po peti destilaciji oziroma izguba alkohola med vsako destilacijo ne sme biti večja od 0,02%.
b) Reagenti:
2 M raztopina kalcijevega hidroksida, ki jo pripravimo tako, da 1 liter vode s temperaturo 60 do 70 °C previdno nalijemo na 120 g gašenega apna (CaO).
c) Priprava vzorca:
Iz mladih in penečih vin odstranimo pretežni del mehurčkov CO(2) s stresanjem 250 do 300 ml vina v 500 ml bučki.
d) Postopek:
V 200 ml merilno bučko nalijemo vino do oznake in zabeležimo temperaturo vina. Vino prelijemo v destilacijsko bučko in nastavimo cev za dovod pare. 200 ml merilno bučko speremo štirikrat zaporedoma s po 5 ml vode in te volumne prelijemo v destilacijsko bučko. Dodamo 10 ml kalcijevega hidroksida b.) točka tega člena) in nekaj poroznih vrelnih kamenčkov. Destilat lovimo v 200 ml merilno bučko, ki smo jo uporabili za izmero volumna vina. Pri navadni destilaciji destiliramo do približno tri četrtine začetnega volumna, pri destilaciji z vodno paro pa 198-199 ml destilata. Z destilirano vodo dopolnimo do 200 ml, temperatura destilata se lahko za največ 2 °C razlikuje od začetne temperature vina. Pazljivo premešamo s krožnimi gibi.
Upoštevamo:
Če vino vsebuje posebno veliko količino amonijevih ionov, redestiliramo destilat pod pogoji, ki so opisani v prvem odstavku te točke le s to razliko, da zamenjamo suspenzijo kalcijevega hidroksida z 1 M raztopino žveplove(VI) kisline (H(2)SO(4)), ki jo razredčimo na 10: 100 (vol: vol).
8. člen
(referenčna metoda)
Z referenčno metodo merimo volumenski delež alkohola v destilatu s piknometrom.
a) Aparatura:
Uporablja se standardiziran piknometer, ki je opisan v prvi alinei točke a) 4. člena tega navodila.
b) Postopek:
Izmerimo navidezno gostoto destilata (točka d.) 7. člena tega navodila) pri t °C, kot je opisano v točki c) 4. člena tega navodila. To gostoto označimo z d(t°C).
c) Podajanje rezultatov
– Izračun:
Z uporabo tabele I, ki je v prilogi 2, poiščemo volumski delež alkohola pri 20 °C. V vodoravni vrsti te tabele z ustrezno T (izraženo s celo številko), takoj pod t °C, poiščemo najmanjšo gostoto, ki pa je večja od d(t°C).
Tabelarično razliko pod to gostoto uporabimo za izračun gostote d pri temperaturi T.
V vrsti s temperaturo T poiščemo gostoto d, takoj nad d'' in izračunamo razliko med gostotama d in d''. To razliko delimo s tabelarično razliko, ki se nahaja desno od d'. Kvocient poda decimalni del volumskega deleža alkohola, celo število pa je na vrhu kolone, v kateri se nahaja gostota d''.
Upoštevamo, da je ta temperaturna korekcija lahko vpisana v računalniški program in se lahko izvrši avtomatično.
– Ponovljivost, r: r = 0,10%.
– Obnovljivost, R: R = 0,19%.
9. člen
(običajne metode)
1. HIDROMETRIJA
a) Aparature:
– Alkoholometer.
– Termometer, umerjen v stopinjah in z razdelki 0,1 °C, od 0 °C do 40 °C, z natančnostjo 1/20 stopinje (0,05 °C).
– Merilni cilinder s premerom 36 mm in višino 320 mm, postavljen navpično s pomočjo izravnalnih vijakov.
b) Postopek:
Destilat (točka d) 7. člena tega navodiloa) nalijemo v merilni cilinder. Cilinder mora biti postavljen navpično. Vstavimo termometer in alkoholometer. Temperaturo odčitamo po enominutnem mešanju za izenačevanje temperature v merilnem cilindru, termometru, alkoholometru in destilatu. Odstranimo termometer in po preteku ene minute odčitamo navidezno koncentracijo alkohola. Odčitamo vsaj trikrat s pomočjo povečevalnega stekla. Navidezno vsebnost alkohola, odčitano pri temperaturi t °C, korigiramo na vpliv temperature s pomočjo tabele II, ki je v prilogi 2.
Temperatura tekočine se sme od temperature prostora razlikovati največ za 5 °C.
2. DENZIMETRIJA Z UPORABO HIDROSTATSKE TEHTNICE
a) Aparature:
Uporabljamo hidrostatsko tehtnico (točka 2.a) 5. člena tega navodila).
b) Postopek:
Navidezno gostoto destilata pri temperaturi t °C izmerimo, kot je opisano v točki 2.b) 2. člena tega navodila.
c) Podajanje rezultatov
Poiščemo volumski delež alkohola pri 20 °C z uporabo metode, opisane v drugi alinei točke 2.b) 5. člena tega navodila in z uporabo tabele I, ki je v prilogi 2, če je plovec iz Pyrex stekla ali z uporabo tabele III, ki je v prilogi 2, če je plovec iz navadnega stekla.
3. REFRAKTOMETRIJA
a) Aparature:
Refraktometer, ki omogoča merjenje lomnega količnika v območju od 1,330 do 1,346.
Odvisno od opreme lahko merimo:
– pri 20 °C s primernim instrumentom,
– ali pri sobni temperaturi t °C z uporabo instrumenta, opremljenega s termometrom, ki omogoča merjenje temperature na 0,05 °C natančno. Tabela s temperaturno korekcijo je takemu instrumentu priložena.
b) Postopek:
Lomni količnik alkoholnega destilata, pridobljenega kot je opisano v točki c) 7. člena tega navodila, izmerimo s pomočjo navodil, ki so priložena uporabljenemu instrumentu.
c) Podajanje rezultatov:
Z uporabo tabele IV, ki je v prilogi 2 odčitamo volumenski delež alkohola, ki ustreza izmerjenemu lomnemu količniku pri 20 °C.
Opomba: Tabela IV, ki je v prilogi 2, podaja volumenski delež alkohola glede na lomni količnik tako za zmesi vode in alkohola, kot za alkoholne destilate. V primeru alkoholnih destilatov moramo upoštevati prisotnost nečistoč (pretežno višjih alkoholov). Prisotnost metanola zmanjšuje lomni količnik in s tem volumenski delež alkohola.
III. Določanje skupnega suhega ekstrakta
10. člen
(splošno)
Skupni suhi ekstrakt ali skupna suha snov vključuje vse snovi, ki pri določenih fizikalnih pogojih niso hlapne. Ti fizikalni pogoji morajo biti taki, da se snovi, ki tvorijo ekstrakt, med testom čim manj spremenijo.
Ekstrakt brez sladkorja je razlika med skupnim suhim ekstraktom in skupnimi sladkorji.
Reducirani ekstrakt je razlika med skupnim suhim ekstraktom in skupnimi sladkorji, nad koncentracijo 1 g/l, kalijevim sulfatom, nad koncentracijo 1 g/l, prisotnim manitolom ali katerokoli kemično substanco, ki je lahko bila dodana vinu.
Preostali (rezidualni) ekstrakt je sladkorja prosti ekstrakt, zmanjšan za nehlapne kisline, izražene kot vinska kislina.
Ekstrakt se izraža v gramih na liter in mora biti določen na 0,5 g natančno.
Princip metode merjenja z denzimetrom:
Skupni suhi ekstrakt je izračunan indirektno iz relativne gostote mošta, za vino pa iz relativne gostote dealkoholiziranega vina. Ta suhi ekstrakt predstavlja količino saharoze, ki ob raztapljanju v litru vode da raztopino z enako relativno gostoto, kot jo ima mošt ali dealkoholizirano vino. Ta količina je prikazana v tabeli I, ki je v prilogi 3 objavljena skupaj s tem navodilom, kot njegov sestavni del.
Relativno gostoto (20/20) dealkoholiziranega vina d(r20)(na 20) izračunamo s pomočjo formule:
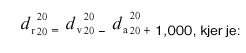
d(v20)(na 20) = relativna gostota vina pri 20 °C (korigirana na hlapne kisline),
d(a20)(na 20) = relativna gostota alkoholnega destilata pri 20 °C mešanice voda-alkohol z enakim volumskim deležem alkohola kot vino,
d(r20)(na 20) lahko izračunamo tudi iz gostot pri 20 °C, gostote vina d(v) in gostote alkoholnega destilata d(a) mešanice voda-alkohol z enakim volumskim deležem alkohola kot vino.
kjer se koeficient 1,0018 približuje 1 ko je d(v) pod 1,05, kar je tudi najbolj pogosto.
Pred izračunom je potrebna korekcija relativne gostote ali gostote vina na vpliv hlapnih kislin z naslednjo formulo:
kjer so a hlapne kisline izražene v miliekvivalentih na liter.
Za izračun skupnega suhega ekstrakta v g/l se uporablja tabela I, ki je v prilogi 3, ki podaja skupni suhi ekstrakt kot funkcijo relativne gostote d(r) (20/20) dealkoholiziranega vina ali relativne specifične
mase mošta d(20°C)(na 20°C).
Skupni suhi ekstrakt se izraža v g/l na eno decimalno mesto natančno.
IV. Določanje skupnih kislin
11. člen
(splošno)
Skupne kisline vina so vsota titracijskih kislin, dobljenih s titracijo do pH 7 s standardno raztopino baze.
Ogljikov dioksid k skupnim kislinam ni vštet.
a) Princip metode za določanje skupnih kislin je potenciometrična titracija ali titracija z indikatorjem bromtimol modro ter primerjava z barvnim standardom končne točke.
b) Reagenti, ki jih uporabljamo pri določanju skupnih kislin:
1. Puferska raztopina pH 7,00:
– kalijev dihidrogen fosfat (KH(2)PO(4)): 107,3 g;
– 1 M raztopina natrijevega hidroksida (NaOH): 500 ml;
– destilirana voda: do 1000 ml.
Lahko uporabimo že pripravljene komercialne puferske raztopine.
2. 0,1 M raztopina natrijevega hidroksida (NaOH);
3. Raztopina indikatorja bromtimol modro s koncentracijo 4 g/l:
– bromtimol modro (C(27)H(28)Br(2)O(5)S): 4 g;
– nevtralni etanol, 96%: 200 ml.
Spojini raztopimo in dodamo:
– degazirano vodo (brez CO(2)): 200 ml;
– 1 M raztopino natrijevega hidroksida, do pojava modro-zelene barve (pH 7): približno 7,5 ml.
Dopolnimo z destilirano vodo do 1000 ml.
c) Aparature in pribor:
– Vodna vakuumska črpalka;
– 500 ml vakuumska bučka;
– Potenciometer s skalo v pH enotah in elektrodi. Steklena elektroda mora biti shranjena v destilirani vodi. Kalomelova elektroda mora biti shranjena v nasičeni raztopini kalijevega klorida. Najpogosteje se uporablja kombinirana elektroda, ki mora biti shranjena v destilirani vodi;
– Merilni valji: za vino 50 ml, za rektificiran koncentriran mošt pa 100 ml.
d) Postopek:
1. Priprava vzorcev:
– Vino:
Izganjanje ogljikovega dioksida: v vakuumsko bučko nalijemo približno 50 ml vina in priključimo na vodno vakuumsko črpalko za eno do dve minuti, vmes pa ves čas mešamo.
– Rektificiran koncentriran mošt:
V 500 ml bučko zatehtamo točno 200 g rektificiranega koncentriranega mošta in razredčimo do značke z vodo. Homogeniziramo.
2. Potenciometrična titracija
– Kalibracija pH metra: pH meter mora biti umerjen na 20 °C, po navodilih proizvajalca, s puferno raztopino pH 7,00 pri 20 °C.
– Metoda merjenja: V merilni valj (četrta alinea c) točke tega člena) nalijemo točno določeno količino vzorca, to je 10 ml vina oziroma 50 ml rektificiranega mošta, pripravljenega kot je opisano v točki d) tega člena. Dodamo približno 10 ml destilirane vode in titriramo z 0,1 M raztopino natrijevega hidroksida (točka b) 2. tega člena) iz birete, dokler ni pH enak 7 pri 20 °C. Med stalnim mešanjem počasi dodajamo bazo. Porabo dodane 0,1 M raztopine natrijevega hidroksida označimo z n ml.
3. Titracija v prisotnosti indikatorja (bromtimol modro)
– Predhoden test: določitev barve v ekvivalentni točki:
V merilni valj (četrta alinea c) točke tega člena) nalijemo 25 ml prevrete destilirane vode, 1 ml raztopine bromtimol modro (prva alinea točke b) 3. tega člena) in določen volumen vzorca, pripravljenega kot je opisano v točki d) tega člena (10 ml pri analizi vina oziroma 50 ml pri analizi rektificiranega koncentriranega mošta). Dodamo 0,1 M raztopino natrijevega hidroksida (točka b) 2. tega člena), dokler se barva ne spremeni v modro-zeleno. Potem dodamo 5 ml puferske raztopine pH 7,0 (točka b) 1. tega člena).
– Titracija: V merilni valj (četrta alinea c) točke tega člena) nalijemo 30 ml prevrete destilirane vode, 1 ml raztopine bromtimol modrega in znan volumen vzorca (vino 10 ml, rektificiran koncentriran mošt 50 ml), pripravljenega kot je opisano v točki d) 1. tega člena. Dodamo 0,1 M raztopino natrijevega hidroksida (točka b) 2. tega člena), dokler ne dosežemo enakega barvnega odtenka kot v predhodnem testu (prva alinea te točke). Porabo dodane 0,1 M raztopine natrijevega hidroksida označimo z n ml.
e) Podajanje rezultatov:
1. Izračun:
– Vino:
Skupne kisline, izražene v miliekvivalentih na liter (A), izračunamo: A = 10×n; Rezultat podamo na eno decimalno mesto natančno.
Skupne kisline, izražene v gramih vinske kisline na liter (A'), izračunamo: A' = 0,075 A; Rezultat podamo na eno decimalno mesto natančno.
– Rektificiran koncentriran mošt:
Skupne kisline, izražene v miliekvivalentih na kilogram rektificiranega koncentriranega mošta, (A) izračunamo:
A = 5 x n;
Skupne kisline, izražene v miliekvivalentih na kilogram skupnih sladkorjev (A), izračunamo:
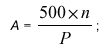
kjer je P masni delež ali masni odstotek v% skupnih sladkorjev. Rezultat podamo na eno decimalno mesto natančno.
2. Ponovljivost (r) pri titraciji z indikatorjem:
Za bela, rdečkasta in rdeča vina:
r = 0,9 mekv / l,
r = 0,07 g vinske kisline / list
3. Reproducibilnost (R) pri titraciji z indikatorjem (točka d) 3.) tega člena:
Za bela in rdečkasta vina:
R = 3,6 mekv / l,
R = 0,3 g vinske kisline / l;
Za rdeča vina:
R = 5,1 mekv / l,
R = 0,4 g vinske kisline / l.
V. Določanje hlapnih kislin
12. člen
(splošno)
Hlapne kisline predstavljajo homologi ocetne kisline, ki se v vinu nahajajo v prosti obliki ali pa v obliki soli.
a) Princip metode: titracija hlapnih kislin, ki jih iz vina ločimo s parno destilacijo in titracija destilata.
Ogljikov dioksid predhodno odstranimo iz vina. Kislost prostega in skupnega žveplovega dioksida v destilatu, dobljenem pri teh pogojih, odštejemo od kislosti destilata. V primeru, da je bila vinu dodana sorbinska kislina, odštejemo od hlapne kislosti tudi kislost le-te.
Pozor: Del salicilne kisline, ki se v nekaterih državah uporablja za stabilizacijo vin pred analizo, je prisoten v destilatu. Količina salicilne kisline v destilatu mora biti določena in odšteta od hlapne kislosti. Metoda za določanje salicilne kisline je podana v točki f) tega člena.
b) Reagenti, ki jih uporabljamo pri določanju hlapnih kislin:
– Kristalinična vinska kislina (C(4)H(6)O(6));
– 0,1 M raztopina natrijevega hidroksida (NaOH);
– 1% raztopina fenolftaleina v 96% nevtralnem alkoholu;
– Klorovodikova kislina, HCl (ro (20) = 1,18 do 1,19 g / ml), razredčena 1: 4 (vol: vol);
– 0,005 M raztopina joda (I(2));
– Kristaliničen kalijev jodid (KI);
– Raztopina škroba, 5 g / l;
5 g škroba zmešamo s 500 ml vode. Med stalnim mešanjem segrejemo do vrenja in pustimo vreti 10 minut. Dodamo 200 g natrijevega klorida. Ko se ohladi, razredčimo na 1 liter.
– Nasičena raztopina natrijevega borata (Na(2)B(4)O(7) × 10 H(2)O), koncentracije približno 55 g / l pri 20 °C.
c) Aparature:
1. Aparaturo za destilacijo z vodno paro sestavljajo:
– parni generator (para ne sme vsebovati ogljikovega dioksida),
– bučka s parno cevko,
– destilacijska kolona,
– kondenzator (hladilnik).
Oprema mora zadostovati naslednjim trem testom:
a) V bučko damo 20 ml prevrete vode. Ulovimo 250 ml destilata, dodamo 0,1 ml 0,1 M raztopine natrijevega hidroksida in 2 kapljici raztopine fenolftaleina. Rožnata barva mora biti obstojna najmanj 10 sekund. To pomeni, da para ne vsebuje ogljikovega dioksida.
b) V bučko damo 20 ml 0,1 M raztopine ocetne kisline. Ulovimo 250 ml destilata in titriramo z 0,1 M raztopino natrijevega hidroksida. Volumen porabljene baze mora biti najmanj 19,9 ml. To pomeni, da je predestilirane najmanj 99,5% ocetne kisline.
c) V bučko damo 20 ml 1M raztopine mlečne kisline. Ulovimo 250 ml destilata in titriramo kislino z 0,1 M raztopino natrijevega hidroksida. Volumen porabljenega natrijevega hidroksida mora biti manjši ali enak 1,0 ml, kar pomeni, da se je predestiliralo največ 0,5% mlečne kisline.
Katerakoli aparatura oziroma postopek, ki zadosti tem trem testom, zadosti zahtevam uradnih mednarodnih aparatur oziroma postopkov.
2. Vodna črpalka
3. Vakuumska bučka
d) Postopek
1. Priprava vzorca: odstranitev ogljikovega dioksida;
V vakuumsko bučko nalijemo približno 50 ml vina; s pomočjo vodne črpalke v bučki vzpostavimo vakuum za približno minuto ali dve in medtem neprenehoma stresamo.
2. Destilacija z vodno paro:
V bučko nalijemo 20 ml vina brez ogljikovega dioksida, kot je opisano v prejšnji točki (točka d) 1. tega člena). Dodamo približno 0,5 g vinske kisline (prva alinea točke b) tega člena). Zberemo vsaj 250 ml destilata.
3. Titracija
Titriramo z 0,1 M raztopino natrijevega hidroksida (druga alinea točke b) tega člena) ob dodatku dveh kapljic fenolftaleina (tretja alinea točke b) tega člena) kot indikatorja. Volumen porabljenega natrijevega hidroksida označimo z n ml.
Dodamo 4 kapljice razredčene raztopine klorovodikove kisline (četrta alinea točke b) 4. tega člena), 2 ml škrobovice (sedma alinea točke b) tega člena) in nekaj kristalov kalijevega jodida (šesta alinea točke b) tega člena). Volumen porabljene jodovice označimo kot n' ml.
Dodamo nasičeno raztopino natrijevega borata (osma alinea točke b) tega člena), dokler se rožnato obarvanje ponovno ne pojavi. Skupni žveplov dioksid titriramo z 0,005 M raztopino jodovice (peta alinea točke b) tega člena). Porabljeni volumen označimo z n'' ml.
e) Podajanje rezultatov
1. Računska metoda:
Hlapne kisline, izražene v miliekvivalentih na liter, na eno decimalno mesto natančno, so podane kot:
A = 5 × (n – 0,1×n' – 0,05×n'');
Hlapne kisline, izražene v gramih ocetne kisline na liter, na dve decimalni mesti natančno, so podane kot:
A = 0,300 × (n – 0,1×n' – 0,05×n'');
2. Ponovljivost (repeatability), r:
r = 0,7 mekv/l;
r = 0,04 g ocetne kisline/l.
3. Obnovljivost (reproducibility), R:
R = 1,3 mekv/l;
R = 0,08 g ocetne kisline/l.
4. Vina s prisotno sorbinsko kislino:
Ker se 96% sorbinske kisline predestilira v destilat volumna 250 ml, moramo njeno kislost odšteti od hlapnih kislin. 100 mg sorbinske kisline odgovarja kislosti 0,89 miliekvivalentov oziroma 0,053 g ocetne kisline. Koncentracijo sorbinske kisline v mg/l določimo s pomočjo drugih metod.
f) Določitev salicilne kisline v destilatu hlapnih kislin
1. Princip:
Pri določanju hlapnih kislin in korekciji na žveplov dioksid pokaže prisotnost salicilne kisline vijolično obarvanje po nakisanju raztopine in dodatku železove (III) soli.
Določitev salicilne kisline, ki se ob hlapnih kislinah nahaja v destilatu, poteka v drugem destilatu enakega volumna kot tisti, v katerem smo določili hlapne kisline. V tem destilatu vsebnost salicilne kisline določamo s primerjalno kolorimetrično metodo. Koncentracijo odštejemo od hlapnih kislin destilata.
2. Reagenti, ki jih uporabljamo pri določitvi salicilne kisline v destilatu hlapnih kislin:
– Klorovodikova kislina, HCl (ro (20) = 1,18 do 1,19 g/l),
– 0,1 M raztopina natrijevega tiosulfata (Na(2)S(2)O(3) * 5H(2)O),
– 10% raztopina amonijevega železovega (III) sulfata ((Fe(2)(SO(4))(3) × (NH(4))(2)SO(4) × 24H(2)O)),
– 0,01 M raztopina natrijevega salicilata; raztopina vsebuje 1,60 g/l natrijevega salicilata (NaC(7)H(5)O(3)).
3. Postopek:
– Identifikacija salicilne kisline v destilatu hlapnih kislin:
Takoj po določitvi hlapnih kislin in korekciji na prosti in skupni žveplov dioksid damo v konično bučko 0,5 ml klorovodikove kisline (prva alinea točke f)2. tega člena), 3 ml 0,1 M raztopine natrijevega tiosulfata (druga alinea točke f) 2. tega člena) in 1 ml raztopine amonijevega železovega (III) sulfata (tretja alinea točke f) 2. tega člena). Ob prisotnosti salicilne kisline se pojavi vijolično obarvanje.
– Določitev salicilne kisline:
Na konični bučki označimo nivo destilata. Bučko spraznimo in izplaknemo. V isto bučko damo novih 20 ml vina, parno destiliramo in zberemo destilat do predhodno narejene oznake. Dodamo 0,3 ml klorovodikove kisline (prva alinea točke f) 2. tega člena) in 1 ml raztopine železovega (III) amonijevega sulfata (tretja alinea točke f) 2. tega člena). Vsebina bučke se obarva vijolično.
V konično bučko, identično bučki iz 1. odstavka te točke, damo destilirano vodo do značke. Dodamo 0,3 ml klorovodikove kisline (prva alinea točke f) 2. tega člena) in 1 ml raztopine železovega (III) amonsulfata (tretja alinea točke f) 2. tega člena). Iz birete dodajamo 0,01 M raztopino natrijevega salicilata (četrta alinea točke f) 2. tega člena), dokler raztopina nima enakega barvnega odtenka kot tista z destilatom vina.
Volumen raztopine, ki smo jo dodali iz birete označimo z n'' ml.
– Korekcija hlapnih kislin:
Od volumna n 0,1 M raztopine natrijevega hidroksida, ki smo ga porabili za titracijo hlapnih kislin, odštejemo 0,1 × n''ml.
VI. Določanje nehlapnih kislin
13. člen
(splošno)
Nehlapne kisline predstavljajo razliko med skupnimi in hlapnimi kislinami.
Nehlapne kisline izražamo v:
– miliekvivalentih na liter (mekv/l),
– gramih vinske kisline na liter (g vinske kisline/l).
VII. Določanje pepela
14. člen
(splošno)
Kot pepel so definirani vsi tisti produkti, ki ostanejo po sežigu ostanka, dobljenega po izparevanju vina. Sežig poteka tako, da vsi kationi (razen amonijevega kationa) preidejo v karbonate ali druge brezvodne anorganske soli.
a) Princip metode:
Ekstrakt vina žarimo pri temperaturi med 500 °C in 550 °C do popolne oksidacije organskih sestavin.
b) Aparature:
– vodna kopel,
– tehtnica z natančnostjo 0,1 mg,
– evaporator z vročo ploščo ali infra-rdeč evaporator,
– žarilna peč s kontrolo temperature,
– eksikator,
– platinske posode z ravnim dnom, premera 70 mm in višine 25 mm.
c) Postopek:
V predhodno stehtano platinsko posodo (začetna masa P(0)) odpipetiramo 20 ml vina. Odparimo do suhega na vreli vodni kopeli, ostanek pa segrevamo na vroči plošči pri 200 °C ali na infra-rdečem evaporatorju do začetka pooglenitve. Ko se tvorba dima ustavi, postavimo posodo v žarilno peč, segreto na 525 + / - 25 °C. Po 15 minutah sežiga jo vzamemo iz peči, dodamo 5 ml destilirane vode, odparimo do suhega na vodni kopeli ali pod infrardečim evaporatorjem in ostanek ponovno žarimo pri 525 °C 10 minut.
Če sežig (oksidacija karboniziranih delcev) ni popoln, ponavljamo postopek dodajanja vode, odparevanje vode in sežig.
Ko je pepel bele barve, platinsko posodo ohladimo v eksikatorju ter stehtamo (masa P(1)).
Masa pepela v vzorcu (20 ml) je torej P = P(1) – P(0).
d) Podajanje rezultatov in izračun:
Masa pepela P v gramih na liter se podaja na dve decimalni mesti z izrazom: P = 50×p.
Opomba: Za vina, ki vsebujejo veliko koncentracijo reducirajočih sladkorjev, se priporoča dodatek nekaj kapljic čistega rastlinskega olja v ekstrakt vina (pred prvim žarjenjem, zaradi preprečitve penjenja).
VIII. Določanje pH vrednosti
15. člen
(splošno)
Merimo razliko potencialov med dvema elektrodama, potopljenima v preiskovano tekočino. Potencial ene od elektrod je odvisen od pH vrednosti tekočine, medtem ko ima druga elektroda stalen in znan potencial ter predstavlja referenčno elektrodo.
a) Aparatura:
– pH meter s kalibrirano skalo v pH enotah in možnostjo merjenja na najmanj 0,05 pH enote natančno,
– elektrodi: steklena elektroda; hranimo jo v destilirani vodi,
kalomelova elektroda – referenčna; hranimo v nasičeni raztopini kalijevega klorida,
ali kombinirana elektroda; hranimo jo v destilirani vodi.
b) Reagenti – puferske raztopine:
– Nasičena raztopina kalijevega hidrogentartrata, ki vsebuje najmanj 5,7 g kalijevega hidrogen tartrata na liter (C(4)H(5)KO(6)) pri 20 °C. (Raztopino lahko hranimo do dveh mesecev, če ji dodamo 0,1 g timola na 200 ml).
pH vrednost: 3,57 pri 20 °C,
3,56 pri 25 °C,
3,55 pri 30 °C;
– 0,05 M raztopina kalijevega hidrogenftalata, ki vsebuje 10,211 g kalijevega hidrogenftalata (C(8)H(5)KO(4)) pri 20 °C. (Hranimo jo lahko maksimalno dva meseca).
PH vrednost: 3,999 pri 15 °C,
4,003 pri 20 °C,
4,008 pri 25 °C,
4,015 pri 30 °C;
– Raztopina, ki vsebuje:
monokalijev fosfat, KH(2)PO(4): 3,402 g,
dikalijev fosfat, K(2)HPO(4): 4,354 g,
vodo: do 1000 ml.
(Raztopino lahko hranimo maksimalno 2 meseca.)
pH vrednost: 6,90 pri 15 °C,
6,88 pri 20 °C,
6,86 pri 25 °C,
6,85 pri 30 °C;
Opomba: Možna je uporaba že pripravljenih puferskih raztopin.
c) Postopek:
1. Priprava vzorca za analizo:
– Mošte in vina merimo direktno.
– Rektificirane koncentrirane mošte razredčimo z vodo do masnega deleža 25 + / - 5% (m/m) skupnih sladkorjev (25 °Brix). Če je P koncentracija (m/m) v odstotkih skupnih sladkorjev, v rektificiranem koncentriranem moštu zatehtamo: 2500/P in razredčimo do 100 g z vodo. Prevodnost vode mora biti pod 2 mikrosiemensa na cm (mikroS/cm).
2. Nastavljanje aparata na ničlo:
Nastavljanje na ničlo se izvede pred merjenjem po navodilih, ki so priložena aparaturi.
3. Umerjanje pH metra:
pH meter umerimo pri 20 °C s puferno raztopino pH = 6,88 in 3,57, ki imata prav tako 20 °C. Za preverjanje skale uporabimo puferno raztopino pH = 4,00 pri 20 °C.
4. Meritev:
Elektrodo potopimo v vzorec s temperaturo med 20 do 25 °C oziroma čim bližje 20 °C. pH vrednost direktno odčitamo na skali. Vsak vzorec izmerimo vsaj dvakrat. Končni rezultat naj bo aritmetična sredina obeh meritev.
d) Podajanje rezultata:
pH vrednost mošta, vina ali 25% (m/m) (25 °Brix) raztopine rektificiranega koncentriranega mošta podamo na dve decimalni mesti natančno.
IX. Določanje reducirajočih sladkorjev
16. člen
(splošno)
Reducirajoči sladkorji so vsi sladkorji, ki imajo keto in aldehidne funkcionalne skupine. Njihovo določevanje je povezano z redukcijo alkalne raztopine bakrove soli (II).
Principi metod:
1. Čiščenje:
– Referenčna metoda: Po nevtralizaciji in odstranitvi alkohola vino spustimo skozi ionsko izmenjalno kolono, v kateri se njegovi anioni izmenjajo z acetatnimi anioni, temu pa sledi čiščenje z nevtralnim svinčevim acetatom.
– Običajne metode: Vino čistimo z enim od naslednjih reagentov: nevtralni svinčev acetat, cink-2 heksacianoferat.
2. Določitev:
Očiščeno vino ali mošt reagira z določeno količino alkalne raztopine bakrove soli(II), prebitek bakrovih ionov pa določimo jodometrično.
17. člen
(čiščenje vina)
Koncentracija reducirajočih sladkorjev v vzorcu, ki ga analiziramo, mora biti med 0,5 in 5 g/l. Suhih vin med čiščenjem ne razredčujemo, sladka vina moramo razredčiti med čiščenjem tako, da je vsebnost sladkorja v mejah, ki jih predpisuje naslednja tabela:
-----------------------------------------------------------------
Opis vzorca Koncentracija Relativna Razredčitev
sladkorja (g/l) gostota (%)
-----------------------------------------------------------------
mošti in mistele nad 125 nad 1,038 1
-----------------------------------------------------------------
sladka vina 25 do 125 1,005 do 1,038 4
-----------------------------------------------------------------
polsladka vina 5 do 25 0,997 do 1,005 20
-----------------------------------------------------------------
suha vina pod 5 pod 0,997 brez
razredčenja
-----------------------------------------------------------------
1. REFERENČNA METODA
a) Reagenti:
– 1 M raztopina klorovodikove kisline, HCl,
– 1 M raztopina natrijevega hidroksida, NaOH,
– 4 M raztopina ocetne kisline, CH(3)COOH,
– 2 M raztopina natrijevega hidroksida, NaOH.
– Anionska izmenjalna smola (Dowex 3 (20-50 mesh) ali podobno)
Priprava anionske izmenjalne kolone:
Majhen zamašek iz steklene volne in 15 ml anionske izmenjalne smole (peta alinea točke 1.a) tega člena) damo na dno birete. Pred uporabo smolo dvakrat regeneriramo tako, da jo izmenično prelijemo z 1 M raztopino klorovodikove kisline (prva alinea točke 1.a) tega člena) in 1 M raztopino natrijevega hidroksida (druga alinea točke 1.a) tega člena). Po spiranju s 50 ml destilirane vode, prenesemo smolo v čašo, dodamo 50 ml 4 M raztopine ocetne kisline (tretja alinea točke 1.a) tega člena) in mešamo pet minut. Ponovno napolnimo bireto s smolo in skoznjo prelijemo 100 ml 4 M raztopine ocetne kisline.
Opomba: Zaželjeno je, da je zaloga smole spravljena v steklenici, ki je napolnjena z 4 M raztopino ocetne kisline.
Kolono spiramo z destilirano vodo, dokler eluat ni nevtralen.
Regeneracija smole:
Skozi kolono nalijemo 150 ml 2 M raztopine natrijevega hidroksida, da odstranimo kisline in večino barvil, ki se vežejo na smolo. Speremo s 100 ml vode in nato nalijemo 100 ml 4 M raztopine ocetne kisline. Kolono spiramo z destilirano vodo, dokler eluat ni nevtralen.
– Nevtralna raztopina svinčevega acetata (približno nasičena):
nevtralni svinčev acetat (Pb(CH(3)COO)(2) × 3H(2)O): 250 g,
zelo vroča voda: do 500 ml,
mešamo, dokler se popolnoma ne raztopi.
– Kalcijev karbonat (CaCO(3)).
b) Postopek:
– Suha vina:
50 ml vina nalijemo v čašo s premerom približno 10 do 12 cm, dodamo 1/2 (n – 0,5) ml 1 M raztopine natrijevega hidroksida (druga alinea točke 1.a) tega člena), (n je volumen 0,1 M raztopine natrijevega hidroksida, ki ga porabimo za titracijo skupnih kislin v 10 ml vina) in odparevamo v vodni kopeli v toku toplega zraka, dokler se volumen tekočine ne zmanjša na približno 20 ml. To tekočino spustimo skozi anionsko izmenjevalno smolo v acetatni obliki (peta alinea točke 1.a) tega člena) po 3 ml vsake 2 minuti. Eluat zbiramo v 100 ml bučko. Posodo in kolono speremo šestkrat s po 10 ml destilirane vode. Med mešanjem dodamo 2,5 ml nasičene raztopine svinčevega acetata (šesta alinea točke 1.a) tega člena) in 0,5 g kalcijevega karbonata (sedma alinea točke 1.a) tega člena) v eluat, nato večkrat stresemo in pustimo stati najmanj 15 minut. Bučko dopolnimo do oznake in filtriramo. 1 ml tega filtrata ustreza 0,5 ml vina.
– Mošti, mistele, sladka in polsladka vina:
Razredčitve so podane kot vodilo:
1. mošti in mistele: pripravimo 10% raztopino vzorca in za analizo vzamemo 10 ml tega razredčenega vzorca;
2. sladka vina, obogatena ali ne, z relativno gostoto med 1,005 in 1,038: pripravimo 20% raztopino vzorca in vzamemo 20 ml razredčenega vzorca;
3. polsladka vina z relativno gostoto pri 20 °C med 0,997 in 1,005: vzamemo 20 ml nerazredčenega vzorca.
Zgoraj omenjene volumne vina ali mošta spuščamo skozi anionsko izmenjalno kolono v acetatni obliki, po 3 ml vsake 2 minuti. Eluat zbiramo v 100 ml bučki, kolono spiramo z vodo, dokler ne dobimo približno 90 ml eluata. V eluat dodamo 0,5 g kalcijevega karbonata in 1 ml nasičene raztopine svinčevega acetata. Zmešamo in pustimo stati 15 minut, občasno premešamo. Nato dopolnimo z vodo do oznake in filtriramo.
V primeru iz zgornjih točk 1., 2. in 3. razredčitve ustrezajo naslednjim količinam:
1. 1 ml filtrata ustreza 0,01 ml mošta ali mistele,
2. 1 ml filtrata ustreza 0,04 ml sladkega vina,
3. 1 ml filtrata ustreza 0,20 ml polsladkega vina.
2. OBIČAJNE METODE
A. ČIŠČENJE Z NEVTRALNIM SVINČEVIM ACETATOM
a) Reagenti:
– Raztopina nevtralnega svinčevega acetata (približno nasičena; glej šesto alineo točke 1.a) tega člena);
– Kalcijev karbonat.
b) Postopek:
– Suha vina:
50 ml vina nalijemo v 100 ml merilno bučko, dodamo 1/2 (n – 0,5) ml 1 M raztopine natrijevega hidroksida (druga alinea točke 1.a) tega člena; kjer je nvolumen 0,1 M raztopine natrijevega hidroksida porabljen za določitev skupnih kislin v 10 ml vina). Med mešanjem dodamo 2,5 ml nasičene raztopine svinčevega acetata (šesta alinea točke 1.a) tega člena) in 0,5 g kalcijevega karbonata (sedma alinea točka 1.a) tega člena). Bučko pretresemo in pustimo stati vsaj 15 minut. Dopolnimo do oznake z vodo in filtriramo. 1 ml filtrata ustreza 0,5 ml vina.
– Mošti, mistele, sladka in polsladka vina:
Razredčitve so podane kot vodilo:
1. mošti in mistele: pripravimo 10% raztopino vzorca in za analizo vzamemo 10 ml tega razredčenega vzorca;
2. sladka vina, obogatena ali ne, z gostoto med 1,005 in 1,038: pripravimo 20% raztopino vzorca in vzamemo 20 ml razredčenega vzorca;
3. polsladka vina z gostoto pri 20 °C med 0,997 in 1,005: vzamemo 20 ml nerazredčenega vzorca.
Dodamo 0,5 g CaCO(3), približno 60 ml vode in 0,5, 1 ali 2 ml nasičene raztopine svinčevega acetata. Premešamo, pustimo stati vsaj 15 minut, občasno mešamo. Dopolnimo do oznake z vodo in nato filtriramo.
V primeru iz zgornjih točk 1., 2. in 3. razredčitve ustrezajo naslednjim količinam:
1. 1 ml filtrata ustreza 0,01 mošta ali mistele,
2. 1 ml filtrata ustreza 0,04 ml sladkega vina,
3. 1 ml filtrata ustreza 0,20 ml polsladkega vina.
B. ČIŠČENJE S CINKOVIM-2 HEKSACIANOFERATOM
Ta postopek uporabljamo le za bela vina, rahlo obarvana sladka vina in mošte.
a) Reagenti:
– Raztopina I: kalijev 2-heksacianoferat:
Kalijev 2-heksacianoferat (K(4)Fe(CN)(6) × 3H(2)O), 150 g; dopolnimo z vodo do 1000 ml.
– Raztopina II: cinkov sulfat:
Cinkov sulfat, ZnSO(4) × 7H(2)O, 300 g; dopolnimo z vodo do 1000 ml.
b) Postopek:
V 1000 ml bučko nalijemo vzorec vina (ali mošta ali mistele); razredčitve so podane kot vodilo:
1. mošti in mistele: pripravimo 10% raztopino vzorca in za analizo vzamemo 10 ml razredčenega vzorca;
2. sladka vina, obogatena ali ne, z relativno gostoto med 1,005 in 1,038: pripravimo 20% raztopino vzorca in vzamemo 20 ml razredčenega vzorca;
3. polsladka vina z relativno gostoto med 0,997 in 1,005: vzamemo 20 ml nerazredčenega vzorca;
4. suha vina: 50 ml nerazredčenega vzorca.
Dodamo 5 ml raztopine I, kalijev 2-heksacianoferat (1 alinea točke 2.B.a) tega člena) in 5 ml raztopine II, cinkov sulfat (druga alinea točke 2.B.a) tega člena). Premešamo, dopolnimo z vodo do oznake, pustimo stati 10 minut in nato filtriramo.
V primeru iz zgornjih točk 1., 2., 3. in 4. razredčitve ustrezajo naslednjim količinam:
1. 1 ml filtrata ustreza 0,01 ml mošta ali mistele,
2. 1 ml filtrata ustreza 0,04 ml sladkega vina,
3. 1 ml filtrata ustreza 0,20 ml polsladkega vina,
4. 1 ml filtrata ustreza 0,50 ml suhega vina.
18. člen
(določanje sladkorjev)
a) Reagenti:
– Alkalna raztopina bakrove soli:
bakrov sulfat (CuSO(4) × 5H(2)O): 25 g;
citronska kislina (C(6)H(8)O(7) × H(2)O): 50 g;
natrijev karbonat (Na(2)CO(3) × 10H(2)O): 388 g;
voda: do 1000 ml.
Bakrov sulfat raztopimo v 100 ml vode, citronsko kislino v 300 ml vode in natrijev karbonat v 300 do 400 ml vroče vode. Zmešamo raztopini citronske kisline in natrijevega karbonata. Dodamo raztopino bakrovega sulfata in dopolnimo do litra.
– 30% raztopina kalijevega jodida:
kalijev jodid (KI): 30 g;
voda: do 100 ml.
Raztopino hranimo v temni steklenici.
– 25% raztopina žveplove (VI) kisline:
koncentrirana žveplena kislina (H(2)SO(4)), ro = 1,84 g/ml: 25 g;
voda: do 100 ml.
Kislino počasi dodajamo k vodi, počakamo da se ohladi in dopolnimo do 100 ml z vodo.
– Raztopina škroba, 5 g/l:
5 g škroba zmešamo s približno 500 ml vode. Ob stalnem mešanju segrejemo do vrenja in pustimo vreti 10 minut. Dodamo 200 g natrijevega klorida (NaCl), ohladimo in dopolnimo z vodo do 1 litra.
– 0,1 M raztopina natrijevega tiosulfata;
– Raztopina invertnega sladkorja, 5 g/l (za preverjanje metode določanja);
V 200 ml bučko damo:
čisto suho saharozo (C(12)H(22)O(11)): 4,75 g;
vodo: ca 100 ml;
koncentrirana HCl, ro(20) = od 1,16 do 1,19 g/ml: 5 ml.
Bučko segrevamo na vodni kopeli (temperatura kopeli 60 °C) dokler temperatura raztopine ne doseže 50 °C; to temperaturo vzdržujemo 15 minut. Bučko hladimo na zraku 30 minut, nato jo potopimo v hladno vodo. Raztopino prenesemo v 1 litrsko merilno bučko in dopolnimo do oznake. Ta raztopina je obstojna 1 mesec. Kadar jo uporabljamo, testni alikvot (raztopina vsebuje cca. 0,06 M HCl) nevtraliziramo z raztopino natrijevega hidroksida.
b) Postopek:
V 300 ml erlenmajerici zmešamo 25 ml alkalne raztopine bakrove soli, 15 ml vode in 10 ml očiščene raztopine vzorca. Ta volumen raztopine ne sme vsebovati več kot 60 mg invertnega sladkorja. Dodamo nekaj vrelnih kamenčkov, na erlenmajerico pritrdimo hladilnik in v dveh minutah segrejemo do vrenja. Mešanica mora vreti točno 10 minut. Erlenmajerico ohladimo pod mrzlo tekočo vodo. Ko je popolnoma hladna, dodamo 10 ml 30% raztopine kalijevega jodida (druga alinea točke a) tega člena), 25 ml 25% raztopine žveplove kisline (tretja alinea točke a) tega člena) in 2 ml raztopine škrobovice (četrta alinea točke a) tega člena). Titriramo z 0,1 M raztopino natrijevega tiosulfata (peta alinea točke a) tega člena), število porabljenih mililitrov označimo z n. Za slepo titracijo nadomestimo 10 ml raztopine vzorca z 10 ml destilirane vode, število porabljenih ml natrijevega-tiosulfata za slepo titracijo označimo z n'.
c) Podajanje rezultatov:
1. Izračuni:
Masa reducirajočih sladkorjev, izražena kot invertni sladkor, vsebovana v testnem vzorcu, je podana v tabeli I, ki je v prilogi 4 objavljena skupaj s tem navodilom, kot njegov sestavni del, kot funkcija števila (n' – n) porabljenih ml Na-tiosulfata.
Maso reducirajočih sladkorjev podajamo v g invertnega sladkorja v 1 litru, na eno decimalno mesto in pri tem upoštevamo razredčitev med čiščenjem in volumen testnega vzorca.
2. Ponovljivost (repeatability), r: r = 0,015x(i),
kjer je x(i) koncentracija invertnega sladkorja v g/l v vzorcu.
3. Obnovljivost (reproducibility), R: R = 0,058 x(i),
kjer je x(i) koncentracija invertnega sladkorja v g/l v vzorcu.
X. Določanje prostega in skupnega žveplovega dioksida
19. člen
(splošno)
Prosti žveplov dioksid je definiran kot žveplov dioksid, ki je prisoten v moštu ali vinu v naslednjih oblikah: H(2)SO(3), HSO(3)(na -).
Ravnotežje med tema oblikama je funkcija pH vrednosti in temperature:
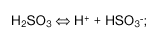
H(2)SO(3) predstavlja molekularno obliko žveplovega dioksida.
Skupni žveplov dioksid je definiran kot vsota različnih oblik žveplovega dioksida, prisotnih v vinu v prosti obliki ali vezanih s sestavinami vina.
Principi metod:
1. Referenčna metoda:
Za vina in mošte:
Žveplov dioksid odstranimo iz vina s tokom zraka oziroma dušika; fiksiramo in oksidiramo ga tako, da mehurčke vodimo skozi razredčeno in nevtralno raztopino vodikovega peroksida. Nastalo žveplovo(VI) kislino določimo s titracijo s standardno raztopino natrijevega hidroksida.
Prosti žveplov dioksid odstranimo iz vina z vzdrževanjem nizke temperature (10 °C).
Skupni žveplov dioksid odstranimo iz vina s segrevanjem na visoke temperature (približno 100 °C).
Za rektificirane koncentrirane mošte:
Skupni žveplov dioksid odstranimo iz predhodno razredčenega rektificiranega koncentriranega mošta s segrevanjem na visoke temperature (približno 100 °C).
2. Hitra metoda določanja
Za vina in mošte:
Prosti žveplov dioksid določimo z direktno jodometrično titracijo.
Vezani žveplov dioksid določimo z jodometrično titracijo po alkalni hidrolizi. Ta rezultat prištejemo k rezultatu za prosti žveplov dioksid, vsota pa predstavlja skupni žveplov dioksid.
20. člen
(referenčna metoda)
a) Aparatura:
– Aparatura, ki jo uporabljamo, mora ustrezati sliki 2. Posebno pozornost je potrebno nameniti kondenzatorju (hladilniku).
Slika 2: Dimenzije so podane v milimetrih. Notranji premeri štirih koncentričnih cevk, ki ustrezajo kondenzatorju (hladilniku) so: 45, 34, 27 in 10 mm.
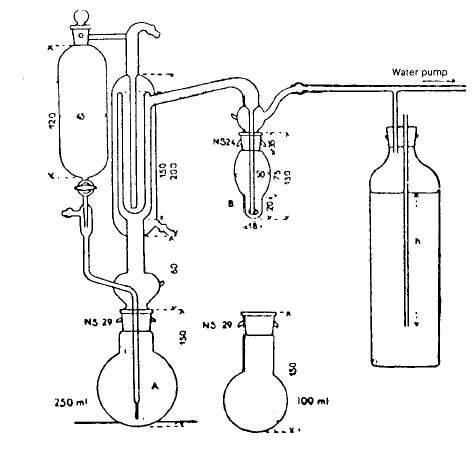
Cevka, po kateri izhaja iz vzorca plin, se v bučki B razširi v kroglico premera 1 cm z dvajsetimi luknjicami premera 0,2 mm okoli največjega obsega. Izjemoma se cevka lahko zaključi s stekleno porozno frito, ki ustvarja veliko število zelo majhnih mehurčkov in zagotavlja dober kontakt med plinsko in tekočo fazo.
Pretok plina skozi aparaturo naj bo približno 40 litrov na uro. Steklenica na desni strani skice je namenjena omejitvi padca tlaka, ki nastane zaradi vodne črpalke, na nivo 20 do 30 cm. Za regulacijo pravilnega podtlaka moramo med bučko in steklenico vstaviti merilec pretoka s pol kapilarno cevko.
– Mikrobireta.
b) Reagenti:
– Fosforna kislina, 85% (H(3)PO(4), ro(20) = 1,71 g/ml);
– Raztopina vodikovega peroksida, 9,1 g H(2)O(2) /liter (trije volumni);
– Indikator:
metil rdeče: 100 mg,
metilen modro: 50 mg,
alkohol, 50%: 100 mg.
– 0,01M raztopina natrijevega hidroksida, NaOH.
c) Postopek
1. Določanje prostega žveplovega dioksida:
Vino moramo imeti spravljeno v polni in zamašeni steklenici pri 20 °C dva dneva pred določanjem. V bučko B damo 2 do 3 ml raztopine vodikovega peroksida (druga alinea točke b) tega člena) in dve kapljici indikatorja. Raztopino vodikovega peroksida nevtraliziramo z 0,01 M raztopino natrijevega hidroksida (četrta alinea točke b) tega člena) in povežemo bučko z aparaturo. V bučko A nalijemo 50 ml vzorca in 15 ml fosforne kisline (prva alinea točke b) tega člena) in jo povežemo z aparaturo. Tok zraka (ali dušika) naj deluje 15 minut. Sproščeni žveplov dioksid se v bučki B oksidira v žveplovo kislino. Bučko B snamemo in titriramo nastalo kislino z 0,01 M raztopino natrijevega hidroksida (četrta alinea točke b) tega člena). Porabljeni volumen za titracijo označimo z n ml.
– Podajanje rezultatov:
Sproščeni žveplov dioksid izrazimo v mg/l na najbližjo celo številko.
Izračun: Prosti žveplov dioksid v mg/l je produkt 6,4n.
2. Določanje skupnega žveplovega dioksida:
V primeru rektificiranega koncentriranega mošta uporabimo raztopino, dobljeno z razredčitvijo vzorca do 40% (m/v) po navodilih iz poglavja IV. (druga alinea točke d)1. 11. člena tega navodiloa. V 250 ml bučko damo 50 ml razredčenega vzorca in 5 ml fosforne kisline (prva alinea točke b) tega člena). Bučko povežemo z aparaturo.
Vina in mošti:
Če ocenjena koncentracija skupnega SO(2) v vzorcu ni večja od 50 mg/l, damo v 250 ml bučko 50 ml vzorca in 15 ml fosforne kisline (prva alinea točke b) tega člena) ter bučko povežemo z aparaturo.
Če ocenjena koncentracija skupnega SO(2) v vzorcu presega 50 mg/l, damo v 250 ml bučko 20 ml vzorca in 5 ml fosforne kisline (prva alinea točke b) tega člena) ter bučko povežemo z aparaturo.
V bučko B damo 2 do 3 ml raztopine vodikovega peroksida (druga alinea točke b) tega člena) in ga nevtraliziramo po postopku iz točke c)1. tega člena. Vino v bučki A segrejemo do vrenja s pomočjo 4 do 5 cm visokega plamena, ki se dotika dna bučke. Bučke ne smemo postaviti na železno ploščo, ampak na disk s približno 30 mm luknjo v sredini. S tem se izognemo pregrevanju iz vina ekstrahiranih snovi, ki se nalagajo na steni bučke.
Med pretokom zraka (ali dušika) vzdržujemo vrenje. V 15 minutah izženemo in oksidiramo ves žveplov dioksid. Nastalo žveplovo kislino titriramo z 0,01 M raztopino natrijevega hidroksida (četrta alinea točke b) tega člena). Porabljeni volumen za titracijo označimo z n ml.
3. Podajanje rezultatov:
– Mošt in vino: Skupni žveplov dioksid izrazimo v mg/l.
– Rektificiran koncentriran mošt: Skupni žveplov dioksid izrazimo v mg/kg skupnega sladkorja.
– Izračun:
Vino:
Skupni žveplov dioksid v miligramih na liter je:
6,4 × n (če koncentracija SO(2) v vzorcu ni večja od 50 mg/l in smo uporabili 50 ml vzorca);
16 × n (če je koncentracija SO(2) v vzorcu večja od 50 mg/l in smo uporabili 20 ml vzorca).
Rektificirani koncentrirani mošti:
Skupni žveplov dioksid v mg/kg skupnega sladkorja je:
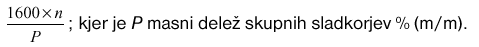
– Ponovljivost (r):
za 50 ml vzorca (koncentracija SO(2) v vzorcu ni večja od 50 mg/l); r = 1 mg/l,
za 20 ml vzorca (koncentracija SO(2) v vzorcu je večja od 50 mg/l); r = 6 mg/l.
– Obnovljivost (R):
za 50 ml vzorca (koncentracija SO(2) v vzorcu ni večja od 50 mg/l); R = 9 mg/l,
za 20 ml vzorca (koncentracija SO(2) v vzorcu je večja od 50 mg/l); R = 15 mg/l.
21. člen
(hitra metoda)
a) Reagenti:
– EDTA komplekson III: dinatrijeva sol etilen diamin tetra ocetne kisline (C(10)H(14)N(2)O(8)Na(2) × 2H(2)O);
– 4 M raztopina natrijevega hidroksida, NaOH, (160 g/l);
– Žveplova(VI) kislina, H(2)SO(4), (ro(20) = 1,84 g/ml), 1: 10 (vol: vol);
– Raztopina škroba 5 g/l; 5 g škroba zmešamo s približno 500 ml vode. Segrevamo do vrenja in med stalnim mešanjem segrejemo do vrenja in vzdržujemo vrenje 10 minut. Dodamo 200 g NaCl. Ohladimo in dopolnimo do enega litra.
– 0,025 M raztopina joda, I(2).
b) Aparatura:
– 500 ml erlenmajerice,
– bireta,
– 1, 2, 5 in 50 ml pipete.
c) Postopek:
1. Prosti žveplov dioksid:
V 500 ml erlenmajerico damo:
– 50 ml vina,
– 5 ml raztopine škroba (četrta alinea točke a) tega člena),
– 30 mg EDTA kompleksona III (prva alinea točke a) tega člena),
– 3 ml raztopine H(2)SO(4), 1/10 (vol: vol) (tretja alinea točke a) tega člena).
Takoj titriramo ob prisotnosti indikatorja škrobovice z 0,025 M raztopino joda do modre barve, ki je obstojna 10 do 15 sekund. Porabo joda označimo z n.
2. Skupni žveplov dioksid:
Dodamo 8 ml 4 M raztopine natrijevega hidroksida (druga alinea točke a) tega člena), mešanico enkrat stresemo in jo pustimo stati 5 minut. Naenkrat in med močnim mešanjem dodamo 10 ml raztopine H(2)SO(4), 1: 10 (v/v; tretja alinea točke a) tega člena). Takoj titriramo z 0,025 M raztopino joda (peta alinea točke 2.a) tega člena), porabljeni volumen označimo z n' ml.
Dodamo 20 ml 4 M raztopine NaOH (druga alinea točke a) tega člena), stresemo in pustimo stati 5 minut. Razredčimo z 200 ml ledeno mrzle vode.
Naenkrat in med močnim mešanjem dodamo vsebino epruvete, v kateri se nahaja 30 ml raztopine H(2)SO(4), 1: 10 (v/v). Prosti žveplov dioksid takoj titriramo z 0,025 M raztopino joda, porabljeni volumen pa označimo z n'' ml.
d) Podajanje rezultatov:
Izračun:
Prosti žveplov dioksid v miligramih na liter: 32 × n
Skupni žveplov dioksid v miligramih na liter: 32 × (n + n' + n'')
Opombe:
– Za rdeča vina z nizko koncentracijo SO(2) je primernejša uporaba raztopine joda, ki je bolj razredčena kot 0,025 M (npr. 0,01 M). V tem primeru zamenjamo v zgornjih formulah faktor 32 z 12,8.
– Za rdeča vina je priporočljiva uporaba osvetlitve od spodaj s snopom rumene luči iz navadne električne žarnice, ki gre skozi raztopino kalijevega kromata ali pa uporabimo natrijevo žarnico. Določanje izvedemo v temni sobi, opazujemo pa transparentnost vina, ki v ekvivalentni točki titracije postane opalescentno.
– Če je koncentracija žveplovega dioksida (H(2)SO(3)) blizu ali pa presega zakonsko dovoljeno mejo, moramo koncentracijo skupnega žveplovega dioksida določiti z referenčno metodo.
– Če je določitev prostega žveplovega dioksida posebno zahtevana, moramo izvesti analizo na vzorcu, ki se je nahajal pri anaerobnih pogojih in temperaturi 20 °C dva dni pred analizo. Tudi analizo izvedemo pri 20 °C.
– Ker jod v kislem mediju oksidira določene substance, moramo to količino joda pravilno ovrednotiti za bolj natančno analizo. V ta namen žveplov dioksid kombiniramo s prebitno količino etanala ali propanala pred izvedbo titracije z jodom. 50 ml vina v 300 ml erlenmajerici dodamo 5 ml raztopine etanala (C(2)H(4)O) s koncentracijo 7 g/l ali 5 ml raztopine propanala (C(3)H(6)O) s koncentracijo 10 g/l.
– Bučko zamašimo in pustimo stati vsaj 30 minut. Dodamo 3 ml raztopine H(2)SO(4), 1: 10 (v/v) in dovolj 0,025 M raztopine joda, da povzročimo spremembo barve škroba. Porabljen volumen raztopine joda označimo z n''' in ga moramo odšteti od n (prosti žveplov dioksid) in od n+ n' + n'' (skupni žveplov dioksid). n''' je ponavadi majhen, od 0,2 do 0,3 ml 0,025 M joda. Če pa je bila vinu dodana askorbinska kislina, je vrednost n''' mnogo večja in obstaja možnost vsaj približnega merjenja količine te kisline s pomočjo vrednosti n''', saj 1 ml 0,025 M raztopine joda oksidira 4,4 mg askorbinske kisline. Z določanjem n''' se dokaj lahko ugotoviti prisotnost rezidualne askorbinske kisline, v koncentracijah nad 20 mg/l v vinih, katerim je bila dodana.
XI. Določanje sladkorja z refraktometrijo
22. člen
(splošno)
a) Princip metode
V tabeli II in tabeli III, ki sta v prilogi 5 objavljeni skupaj s tem navodilom, kot njegov sestavni del, je podan lomni količnik pri 20 °C, izražen bodisi kot absolutna vrednost ali z masnim deležem saharoze, s katerim je mogoče ugotoviti koncentracijo sladkorja v gramih na liter in v gramih na kilogram grozdnega mošta, zgoščenega grozdnega mošta in prečiščenega zgoščenega grozdnega mošta.
b) Aparature
Uporabljamo Abbéjev refraktometer, ki mora imeti skalo, ki prikazuje bodisi:
– masni delež saharoze na 0,1% natančno
– ali lomne količnike na štiri decimalna mesta natančno.
Refraktometer mora imeti termometer, pri katerem skala sega najmanj od 15 °C do 25 °C, in pripravo za kroženje vode, ki omogoča meritve pri temperaturi 20 + / - 5°C.
Navodila za uporabo instrumenta je treba natančno upoštevati, zlasti glede kalibracije in vira svetlobe.
c) Priprava vzorca
– Mošt in zgoščen grozdni mošt
Mošt po potrebi pretočimo skozi štirikrat prepognjeno suho gazo, zavržemo prve kaplje filtrata in nato opravimo določitev filtriranega proizvoda.
– Prečiščen zgoščen grozdni mošt
Glede na koncentracijo uporabimo bodisi samo prečiščen zgoščen grozdni mošt ali raztopino, ki jo dobimo tako, da 200 g prečiščenega koncentriranega mošta do 500 g razredčimo z vodo, pri čemer morajo biti vsa tehtanja natančna.
d) Postopek
Vzorec segrejemo do temperature skoraj 20 °C. Majhen testni vzorec damo na spodnjo prizmo refraktometra, pri čemer pazimo (ker so prizme tesno skupaj), da testni vzorec enakomerno pokriva stekleno površino. Meritev izvedemo v skladu z navodili za uporabo instrumenta.
Odčitamo masni delež saharoze na 0,1% natančno ali lomni količnik na štiri decimalna mesta natančno.
Na istem vzorcu izvedemo najmanj dve določitvi. Zabeležimo temperaturo t °C.
e) Izračun
1. Temperaturna korekcija:
– Instrumenti, ki imajo skalo v masnih deležih saharoze: temperaturni popravek ugotovimo s pomočjo tabele I, ki je v prilogi 5.
– Instrumenti, ki imajo skalo v lomnih količnikih: poiščemo indeks, izmerjen pri t °C v tabeli II ali v tabeli III, ki sta v prilogi 5, da dobimo (stolpec 1) ustrezno vrednost masnega deleža saharoze pri t °C. To vrednost se na podlagi tabele I, ki je v prilogi 5, popravi glede na dejansko temperaturo in izrazi kot koncentracija pri 20 °C.
2. Koncentracija sladkorja v moštu in zgoščenem moštu
Poiščemo masni delež saharoze pri 20 °C v tabeli II, ki je v prilogi 5 in iz iste vrstice odčitamo koncentracijo sladkorja v gramih na liter in gramih na kilogram. Koncentracija sladkorja se izrazi z invertnim sladkorjem na eno decimalno mesto natančno.
3. Koncentracija sladkorja v prečiščenem zgoščenem grozdnem moštu
Poiščemo masni delež saharoze pri 20 °C v tabeli III, ki je v prilogi 5 in iz iste vrstice odčitamo koncentracijo sladkorja v gramih na liter in gramih na kilogram. Koncentracija sladkorja se izrazi z invertnim sladkorjem na eno decimalno mesto natančno.
Če se meritev izvede na prečiščenem zgoščenem grozdnem moštu, rezultat pomnožimo s faktorjem razredčitve.
4. Lomni količnik grozdnega mošta, zgoščenega grozdnega mošta in prečiščenega zgoščenega grozdnega mošta
Poiščemo masni delež saharoze pri 20 °C v tabeli II ali v tabeli III, ki sta v prilogi 5 in iz iste vrstice odčitamo lomni kolilčnik pri 20 °C, ki je izražen na štiri decimalna mesta natančno.
XII. Določanje saharoze
23. člen
(princip metode)
Za kvalitativno testiranje s tankoplastno kromatografijo se saharozo loči od drugih sladkorjev s tankoplastno kromatografijo na plošči, na katero nanesemo sloj celuloze. Sredstvo za razvijanje je sečnina – klorovodikova kislina pri 105 °C.
Za testiranje in določanje s tekočinsko kromatografijo visoke ločljivosti se saharozo separira v koloni s silikagelom, na katerem je kemijsko vezan alkilamil, in detektira z refraktometrijo. Rezultat se ovrednoti glede na eksterni standard, ki je bil analiziran pod istimi pogoji.
Opomba:
Pristnost mošta ali vina se lahko preverja tudi z uporabo metode NMR devterija, ki je opisana pri ugotavljanju obogatitve mošta, prečiščenega zgoščenega mošta in vin.
Za testiranje in določanje saharoze se lahko uporablja tudi kromatografija v plinski fazi.
24. člen
(kvalitativno testiranje s tankoplastno kromatografijo)
a) Oprema
– Kromatografska plošča, na katero nanesemo sloj celuloznega prahu v željeni debelini (npr. MN 300; 20x20).
– Kromatografska posoda (tank).
– Mikrometrična igla ali mikropipeta.
– Pečica z regulacijo do 105 + / - 2°C.
b) Reagenti
– Oglje za razbarvanje.
– Mobilna faza: diklorometan: 100% ocetna kislina (ledocet) (ro(20) = 1,05 g/ml): etanol: metanol: voda, 50: 25: 9: 6: 10.
– Sredstvo za razvijanje:
sečnina: 5 g,
klorovodikova kislina, 2 M: 20 ml,
etanol: 100 ml.
– Referenčne raztopine
glukoza: 35 g,
fruktoza: 35 g,
saharoza: 0,5 g,
destilirana voda: 1000 ml.
c) Postopek
– Priprava vzorca
Če je mošt ali vino močno obarvano, ga razbarvamo z aktivnim ogljem.
Za prečiščeni zgoščeni mošt uporabimo raztopino z masnim deležem sladkorja 25%(m/m) ali 25 °Brix, pripravljeno kot je opisano v drugi alinei točke c)1. 15. člena tega navodila in ga razredčimo z vodo na četrtino koncentracije, tako da 25 ml mošta v merilni bučki dodamo vodo do 100 ml.
– Izdelava kromatograma
2,5 cm od spodnjega roba plošče nanesemo kot črtico 10 mikrol vzorca in 10 mikrol standarda.
Ploščo postavimo v posodo (tank), ki je bila pred tem nasičena s hlapi mobilne faze. Pustimo, da mobilna faza pripotuje do višine 1 cm od gornjega roba plošče. Ploščo vzamemo iz posode in jo osušimo v toku toplega zraka. Postopek ponovimo še dvakrat in ploščo vsakič osušimo. Ploščo enakomerno orosimo s 15 ml razvijalca barve in jo za približno pet minut postavimo v pečico, ogreto na 105 °C.
d) Rezultati
Saharoza in fruktoza se pojavita kot temnomoder madež na belem ozadju, glukoza pa kot manj intenziven zelen madež.
25. člen
(testiranje in določanje s tekočinsko kromatografijo visoke ločljivosti)
Kromatografski pogoji so navedeni samo kot napotek.
a) Oprema
1. Tekočinski kromatograf visoke ločljivosti, ki ima:
– 10 mikrol injektor,
– detektor: diferencialni refraktometer ali interferometer refraktometer,
– kolono s silikagelom, na katerem je kemijsko vezan alkilamil (dolžina 25 cm, notranji premer 4 mm),
– predkolono, napolnjeno z isto fazo,
– pripravo za izoliranje predkolone in analitskih kolon ali za vzdrževanje njihove temperature (30 °C),
– rekorder in po potrebi integrator,
– hitrost pretoka mobilne faze 1 ml/min.
2. Priprava za membransko filtriranje (0,45 mikrom).
b) Reagenti
– Dvakrat destilirana voda.
– Acetonitril (CH(3)CN) HPLC kakovosti.
– Mobilna faza: acetonitril: voda, ki jo pred tem filtriramo z membranskim filtrom (0,45 mikrom), 80:20 (vol: vol).
Iz mobilne faze je treba pred uporabo odstraniti raztopljene pline.
– Standardna raztopina: 1,2 g/l vodne raztopine saharoze. Filtriramo z 0,45 mikrom membranskim filtrom.
c) Postopek
1. Priprava vzorca:
– za vino in mošt: filtriramo z 0,45 mikrom membranskim filtrom,
– za prečiščen zgoščen grozdni mošt: uporabimo raztopino, ki jo dobimo tako, da prečiščen zgoščen mošt razredčimo do 40%, kot je opisano v drugi alinei točke d) 1. 11. člena tega navodila in filtriramo z 0,45 mikrom membranskim filtrom.
2. Kromatografsko določanje
V kromatograf vbrizgamo 10 mikrol standardne raztopine in nato 10 mikrom vzorca, pripravljenega kot je opisano v točki c)1. tega člena. Vbrizgavanje ponovimo v istem vrstnem redu.
Zapišemo kromatogram.
Retencijski čas saharoze je približno 10 minut.
d) Izračuni
Za izračun uporabimo povprečje rezultatov za standardno raztopino in vzorec.
– Za vina in mošt: koncentracije izračunamo v g/l.
– Za prečiščen zgoščen grozdni mošt: C je koncentracija saharoze v g/l v 40% raztopini prečiščenega zgoščenega mošta. Koncentracija saharoze za prečiščen zgoščen grozdni mošt v g/kg je tako 2,5 × C.
e) Izražanje rezultatov
Koncentracija saharoze v vinu, moštu in prečiščenem zgoščenem moštu se izraža v gramih na liter za vina in mošt ter v gramih na kilogram za prečiščen zgoščen grozdni mošt, na eno decimalko natančno.
XIII. Določanje citronske kisline
26. člen
(splošno)
a) Načelo metode
V reakciji, ki jo katalizira encim citrat liaza (CL), se citronska kislina pretvori v oksalacetat in acetat.
V prisotnosti malat dehidrogenaze (MDH) in laktat dehidrogenaze (LDH) se oksaloacetat in njegovi dekarboksilacijski derivati ter piruvat reducirajo z reducirano obliko nikotinamid adenin dinukleotidom (NADH) v L-malat in L-laktat:
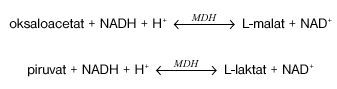
Pri teh reakcijah je količina NADH, ki se oksidira v NAD(na +), sorazmerna s količino prisotnega citrata. Oksidacija NADH se določi z zmanjšanjem absorbcije pri valovni dolžini 340 nm.
b) Reagenti
– Puferska raztopina pH 7,8 (0,51 M raztopina glicilglicina; pH 7,8; Zn(na 2+) (0,6 x 10(na -3) M):
7,13 g glicilglicina raztopimo v približno 70 ml dvakrat destilirane vode. pH uravnamo na 7,8 s približno 13 ml 5 M raztopine natrijevega hidroksida, dodamo 10 ml raztopine cinkovega klorida (80 mg ZnCl(2) v 100 ml H(2)O) in dopolnimo z dvakrat destilirano vodo do 100 ml.
– Raztopina reduciranega nikotinamid adenin dinukleotida (NADH) (približno 6 x 10(na -3) M): 30 mg NADH in 60 mg NaHCO(3) raztopimo v 6 ml dvakrat destilirane vode.
– Raztopina malat dehidrogenaze/laktat dehidrogenaze (MDH/LDH, 0,5 mg MDH/ml, 2,5 g LDH/ml): zmešamo 0,1 ml MDH (5 mg MDH/ml), 0,4 ml raztopine amonijevega sulfata (3,2 M) in 0,5 ml LDH (5 mg/ml). Pri 4 °C je ta suspenzija stabilna najmanj eno leto.
– Citrat liaza (CL, 5 mg protein/ml): 168 mg liofilizata raztopimo v 1 ml ledeno mrzle vode. Ta raztopina ostane stabilna pri 4 °C najmanj en teden in najmanj štiri tedne, če je zmrznjena.
Pred določanjem je priporočljivo preveriti aktivnost encimov.
– Polivinilpolipirolidon (PVPP)
Opomba: Vsi zgoraj omenjeni reagenti so dostopni komercialno.
c) Analitska oprema
– Spektrofotometer, ki omogoča meritve pri 340 nm, t.j. pri valovni dolžini, pri kateri je absorpcija NADH največja.
Lahko pa se uporablja tudi sprektrofotometer z več viri svetlobe, ki omogoča meritve pri 334 nm ali 365 nm.
Ker merimo absolutno absorpcijo (ne uporabljamo umeritvenih krivulj, ampak opravimo standardizacijo z upoštevanjem ekstrakcijskega koeficienta NADH), je treba preveriti lestvice valovnih dolžin in spektralne absorbance opreme.
– Steklene kivete z debelino optične plasti 1 cm ali plastične kivete za enkratno uporabo.
– Mikropipete za dodajanje količin v območju volumnov od 0,02 do 2 ml.
d) Priprava vzorca
Določanje citrata se običajno izvede neposredno v vinu brez predhodne odstranitve pigmentacije (barvil) in brez razredčevanja, če je koncentracija citronske kisline manjša od 400 mg/l. Če je večja, vino razredčimo, dokler koncentracija citrata ni od 20 do 400 mg/l (t.j. od 5 in 80 mikrog citrata v testnem vzorcu).
Rdeča vina vsebujejo veliko fenolnih spojin, zato se priporoča predhodna obdelava s PVPP.
Napravimo vodno suspenzijo približno 0,2 g PVPP in pustimo stati 15 minut. Prefiltriramo s filtrom v obliki lija.
10 ml vina damo v 50 ml erlenmajerico, dodamo vlažen PVPP, ki ga s filtra odstranimo s spatulo. Tresemo dve do tri minute. Filtriramo.
e) Postopek
S spektrofotometrom pri valovni dolžini 340 nm določimo absorpcijo na celicah velikosti 1 cm, pri čemer zrak uporabljamo kot slepi vzorec za referenčno absorpcijo (na poti žarka ni nobene celice). V celico velikosti 1 cm damo naslednje:
-----------------------------------------------------------------
referenčna vzorčna
celica (ml) celica (ml)
-----------------------------------------------------------------
Raztopina 2.1 1,00 1,00
Raztopina 2.2 0,10 0,10
Vzorec za meritev - 0,20
Dvakrat destilirana voda 2,00 1,80
Raztopina 2.3 0,02 0,02
-----------------------------------------------------------------
Mešamo in po približno petih minutah odčitamo absorpcijo raztopine v referenčni in vzorčni celici (A1).
Dodamo:
-----------------------------------------------------------------
Raztopina 2.4 0,02 ml 0,02 ml
-----------------------------------------------------------------
Mešamo in počakamo, dokler se reakcija ne konča (okrog pet minut) in odčitamo absorpcijo raztopin v referenčni in vzorčni celici (A(2)).
Izračunamo razliko absorpcij (A(2) – A(1)) za referenčno celico in celico z vzorcem, in dobimo DeltaAR in DeltaAS.
Na koncu izračunamo razliko med temi razlikami:
DeltaA = DeltaAR – DeltaAS
Opomba: Čas, ki je potreben, da se konča aktivnost encimov, se lahko od ene serije do druge razlikuje. Zgornja vrednost je samo referenca in jo je priporočljivo določiti za vsako posamezno serijo.
f) Izražanje rezultatov
Koncentracija citronske kisline je izražena v miligramih na liter, zaokrožena na najbližje celo število.
1. Postopek izračuna
Splošna formula za izračun koncentracije v mg/l je:
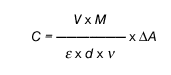
kjer je:
V = volumen testne raztopine v ml (tukaj 3,14 ml)
v = volumen vzorca v ml (tukaj 0,2 ml)
M = molekulska masa, ki jo določamo (tukaj M = 192,1 g/mol za brezvodno citronsko kislino)
d = dolžina optične poti v cm (tukaj 1 cm)
epsilon = absorpcijski koeficient NADH (pri 340 nm, e = 6,3 mmol(na -1) x 1 x cm(na -1),
zato je:
C = 479 x DeltaA
Če je bil vzorec med pripravo razredčen, rezultat pomnožimo s faktorjem razredčitve.
Opomba:
Pri 334 nm: C = 488 x DeltaA (= 6,2 m mol(na -1) x 1 x cm(na -1))
pri 365 nm: C = 887 x DeltaA (= 3,4 m mol(na -1) x 1 x cm(na -1))
2. Ponovljivost (r)
Koncentracija citronske kisline, manjša od 400 mg/l: r = 14 mg/l
Koncentracija citronske kisline, večja od 400 mg/l: r = 28 mg/l
3. Obnovljivost (R)
Koncentracija citronske kisline, manjša od 400 mg/l: R = 39 mg/l
Koncentracija citronske kisline, večja od 400 mg/l: R = 65 mg/l
XIV. Določanje ogljikovega dioksida
27. člen
(princip metod)
– Referenčna metoda
Mirna vina (tlak CO(2) < / = 0,5 x 10(na 5) Pa) (na 1)
Določen volumen vzorca ohladimo na približno 0 °C in zmešamo z zadostno količino natrijevega hidroksida, da dobimo pH vrednost 10 do 11. Titracijo se izvede s kislo raztopino v prisotnosti encima karbonske anhidraze. Koncentracija ogljikovega dioksida se izračuna iz volumna kisline, ki je potreben, da se pH spremeni od 8,6 (hidrogen karbonatna oblika) na 4,0 (ogljikova kislina). Pod istimi pogoji se izvede slepa titracija na razplinjenem vinu, da se upošteva prostornino raztopine natrijevega hidroksida, ki jo sprejmejo vinske kisline.
Peneča in biser vina
Vzorec vina za analizo se ohladi skoraj do ledišča. Ko se odvzame količina, ki se bo uporabila kot slepi vzorec po dekarbonizaciji, je treba preostalo količino v steklenici napraviti alkalno, da se ves ogljikov dioksid veže v obliki Na(2)CO(3). Titracijo se izvede s kislo raztopino v prisotnosti encima karbonske anhidraze. Vsebnost ogljikovega dioksida se izračuna iz prostornine kisle raztopine, ki je potrebna, da se pH spremeni od 8,6 (hidrogen karbonatna oblika) na 4,0 (ogljikova kislina). Pod istimi pogoji se izvede slepa titracija na razplinjenem vinu, da se upošteva volumen natrijevega hidroksida, ki ga sprejmejo vinske kisline.
– Običajna metoda
Peneča in biser vina
Manometrska metoda: z afrometrom se nadtlak ogljikovega dioksida meri neposredno v steklenici.
28. člen
(referenčna metoda)
1. Mirna vina (tlak CO(2) < / = 0,5 x 10(na 5) Pa)
a) Aparature
– Magnetno mešalo
– pH meter
b) Reagenti
– Raztopina natrijevega hidroksida, NaOH, 0,1 M;
– Raztopina žveplove(VI) kisline, H(2)SO(4), 0,05 M;
– Raztopina encima karbonske anhidraze, 1 g/l.
c) Postopek
Vzorec vina ohladimo na približno 0 °C skupaj z 10 ml pipeto, ki je bila uporabljena za odvzem vzorca.
25 ml raztopine natrijevega hidroksida (prva alinea točke 1.b) tega člena) damo v 100 ml čašo; dodamo dve kapljici vodne raztopine ogljikove anhidraze (tretja alinea točke b) tega člena). S pipeto, ohlajeno na 0 °C, dodamo 10 ml vina.
Čašo postavimo na magnetno mešalo, namestimo pH-elektrodo in počasi mešamo.
Ko tekočina doseže sobno temperaturo, jo počasi titriramo z raztopino žveplove(VI) kisline (druga alinea točke 1.b) tega člena) dokler pH ne doseže 8,6. Odčitamo volumen porabljenega titranta na bireti.
Nadaljujemo titracijo z žvepleno kislino, dokler pH ne doseže 4,0.
n je porabljen volumen titranta od vrednosti pH 8,6 do 4,0.
Iz približno 50 ml vzorca vina odstranimo CO(2) tako, da ga tri minute stresamo v vakuumu, pri čemer se bučka greje v vodni kopeli pri približno 25 °C.
Ponovimo zgornji postopek na 10 ml razplinjenega vina.
n’ je porabljen volumen 0,05 M žveplove(VI) kisline.
d) Izražanje rezultatov
1 ml 0,05 M raztopine žveplove(VI) kisline ustreza 4,4 mg CO(2).
Koncentracija CO(2) v gramih na liter vina je podana s formulo:
0,44 x ( n - n' )
Izražena je na dve decimalni mesti natančno. Ovrednotenje rezultatov določa točka 2.e) tega člena.
Opomba: Če vino vsebuje malo CO(2) (CO(2) < 1 g/l), je treba dodati encim karbonsko anhidrazo, ki katalizira hidracijo CO(2).
2. Peneča in biser vina
a) Aparature
– Magnetno mešalo
– pH meter
b) Reagenti
– Natrijev hidroksid, NaOH, 50% (m/m).
– Raztopina žveplove(VI) kisline, H(2)SO(4), 0,05 M.
– Raztopina encima karbonske anhidraze, 1 g/l.
c) Postopek
Označimo nivo vina v steklenici in ga nato ohlajamo, dokler ne začne zmrzovati. Nato pustimo, da se steklenica nekoliko ogreje in jo pri tem stresamo, da ledeni kristalčki izginejo. Hitro odstranimo zamašek in 45 do 50 ml vina damo v merilni valj za slepo titracijo. Točen volumen odvzetega vina v ml se določi tako, da se odčita na valju, ko se ponovno segreje na sobno temperaturo.
Takoj po odvzemu slepega vzorca v steklenico z volumnom 750 ml dodamo 20 ml raztopine natrijevega hidroksida (prva alinea točke 2.b) tega člena).
Počakamo, da se vino segreje na sobno temperaturo.
V 100 ml čašo damo 30 ml prevrete destilirane vode in dve kapljici raztopine encima karbonske anhidraze (tretja alinea točke 2.b) tega člena). Dodamo 10 ml vina, ki smo ga napravili alkalnega. Čašo postavimo na magnetno mešalo, namestimo elektrodo in magnet in počasi mešamo.
Počasi titriramo z raztopino žveplove(VI) kisline (druga alinea točke 2.b) tega člena), dokler pH ne doseže 8,6. Odčitamo volumen porabljenega titranta na bireti.
Počasi nadaljujemo titracijo z žveplovo(VI) kislino, dokler pH ne doseže 4,0.
n je porabljen volumen titranta od vrednosti pH 8,6 do 4,0.
Iz v ml vina, ki smo ga dali na stran za slepo titracijo, odvzamemo CO(2) tako, da ga tri minute stresamo pod vakuumom v bučki, ki se greje v vodni kopeli pri približno 25 °C. Odvzamemo 10 ml razplinjenega vina in dodamo 30 ml prevrete destilirane vode, dodamo dve do tri kaplje raztopine natrijevega hidroksida, da dobimo pH od 10 do 11. Nato izvedemo postopek iz prejšnjega odstavka.
n’ je porabljen volumen 0,05 M žveplove(VI) kisline.
d) Izražanje rezultatov
1 ml 0,05 M raztopine žveplove(VI) kisline ustreza 4,4 mg CO(2).
Izpraznimo steklenico vina, katerega smo napravili alkalnega, in določimo na približno 1 ml natančno začeten volumen vina, tako da do oznake dolijemo vodo. Volumen zabeležimo z V ml.
Koncentracija CO(2) v gramih na liter vina je podana s formulo:
Rezultat se izrazi na dve decimalni mesti natančno.
Ovrednotenje rezultatov določa točka 2.e) tega člena.
e) Ovrednotenje rezultatov
Nadtlak pri 20 °C (P(20)), izražen v pascalih, je podan z naslednjo formulo:
kjer je:
Q = koncentracija CO(2) v g/l vina;
A = volumski delež alkohola pri 20 °C v%;
S = vsebnost sladkorja v vinu v g/l;
Patm = zračni tlak v atmosferi v Pa.
29. člen
(običajna metoda)
1. Peneča in biser vina
a) Aparatura
– Afrometer
Oprema, ki omogoča merjenje nadtlaka v steklenicah s penečim in biser vinom, se imenuje afrometer. Ima različno obliko glede na način, kako je steklenica zamašena (kovinska kronska zaporka, navojna zaporka, plutovinast zamašek ali plastični zamašek.
Afrometer ima skalo v pascalih (Pa), čeprav je bolj praktično kot enoto uporabljati 10(na 5) Pa ali kilopascal (kPa).
Afrometri spadajo v različne razrede. Razred manometrov pomeni natančnost merjenja glede na celotno skalo, izražena kot odstotek (npr. 1000 kPa razred manometrov I pomeni, da se največji tlak 1000 kPa lahko odčita na + / - 10 kPa natančno.) Za natančnost merjenja se priporoča instrumente razreda I.
Afrometre je treba redno pregledovati (najmanj enkrat letno).
b) Postopek
Meritve je treba opraviti na steklenicah, katerih temperatura je konstantna najmanj 24 ur.
Ko predremo zaporko, plutovinast ali plastični zamašek, moramo steklenico močno stresati, dokler tlak ne postane konstanten in lahko odčitamo vrednost.
c) Izražanje rezultatov
Nadtlak pri 20 °C (P(20)) je izražen v pascalih (Pa) ali v kilopascalih (kPa).
Izraziti ga je treba v obliki, ki je skladna z natančnostjo manometra (npr. pri manometrih razreda I s celotno skalo 1000 kPa kot 6,3 x 10(na 5) Pa ali 630 kPa in ne kot 6,33 x 10(na 5) Pa ali 633 kPa).
Če je temperatura, pri kateri se opravi meritev, drugačna od 20 °C, je potreben popravek – izmerjeni tlak se pomnoži s koeficientom:
podanim v tabeli I, ki je v prilogi 6 objavljena skupaj s tem navodilom, kot njegov sestavni del. Na ta način dobimo rezultat pri 20 °C.
30. člen
(razmerje med tlakom in koncetracijo ogljikovega dioksida, ki ga vsebujejo biser vina)
Iz nadtlaka pri 20 °C (P(20)) se absolutni tlak pri 20 °C(Pabs) izračuna po formuli:
Pabs = Patm + P(20)
Koncentracija ogljikovega dioksida v vinu je podana z naslednjim razmerji:
– v litrih CO(2) na liter vina:
- v gramih CO(2) na liter vina:
kjer je A alkoholna stopnja vina pri 20 °C, S je vsebnost sladkorja v vinu v gramih na liter.
XV. Končna določba
31. člen
(veljavnost navodila)
To navodilo začne veljati petnajsti dan po objavi v Uradnem listu Republike Slovenije.
Št. 321-03-42/01-7
Ljubljana, dne 09. maja 2001
mag. Franc BUT l. r.
Minister
za kmetijstvo,
gozdarstvo in prehrano